

As decision-makers in the medical device and therapeutics industries, why do we do what we do? Sure, we want to make money and to some degree, stroke our ego. More likely, our primary driver is that we are wired to help our fellow humans. Our vision is straightforward – we want to improve people’s lives by advancing the standard of care through efficacy and affordability.
The complexity of achieving this “simple” vision is immense, requiring the coordination of hundreds of functional areas. In this blog, we will discuss the sterilization validation and strategies you can use to ensure product safety and effectiveness are preserved.
Explaining Sterilization Validation
The Food and Drug Administration (“FDA”) is tasked with ensuring the safety, efficacy, and security of therapeutics and medical devices. One important aspect of safety is microbial control. As part of an FDA submission, you must provide sterilization validation data confirming microbial control.
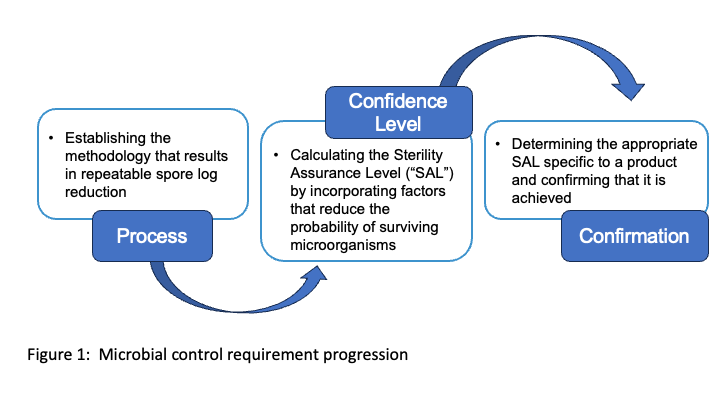
Understanding the progression of the microbial control requirement as you work towards a functionally safe product is important. As shown in Figure 1, there are three aspects to be addressed for the microbial control requirement. Sterilization validation is the confirmation component. We will address the spore log reduction (process) and sterility assurance level (confidence level) aspects in future blogs.
What is Sterilization Validation?
Sterilization validation is a quality control function that is critical to the regulatory review process.
Sterile describes a condition where there are no live microorganisms (such as bacteria, yeast, and viruses).1 It is impossible to ensure sterility 100% of the time. Thus, for regulatory purposes around the globe, sterility is defined according to specific criteria based on the probability of contamination.1 For example, a medical device is considered terminally sterilized when there is a chance of one in a million that it is contaminated with a replicating microorganism.1 This is referred to as having a Sterility Assurance Level (SAL) of 10-6 or SAL6.
This is where sterilization validations come into play. Our experts describe sterilization validation as a way to confirm that specific operating process parameters achieve the intended level of sterility for a particular product. Sterilization validation confirms the effectiveness of the process by checking for the presence of microorganisms on the product in the final packaging configuration after it has gone through the established sterilization cycle.1

Determining the Appropriate Sterilization Validation Strategy
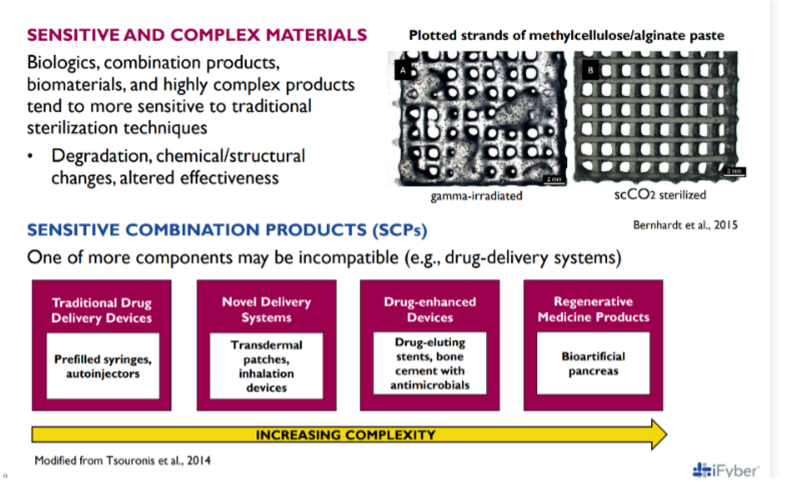
No two products are the same. The complexities of today’s hydrolytically and thermally sensitive materials not only require novel approaches to terminal sterilization, but they also require creative methods of sterilization validation if the utility is to be maintained. Sterilization validation ensures that the selected sterilization process is effective for a given device.
It is important to remember that the FDA is not only concerned about safety. They are also responsible for verifying efficacy. As technologies have advanced and products have become more complex as shown in Figure 2, it is often the case that the materials used are more delicate. Because of this, traditional approaches to sterilization will often not work. The new sterilization methods necessary to preserve product functionality have resulted in alternative sterilization validations being more broadly used.
Following defined standards when performing sterilization validations is necessary for meeting defined regulatory requirements. There are three strategies/approaches that can be used for a sterilization validation study:
- The overkill method,
- The bioburden method, and
- The combined BI/bioburden method.
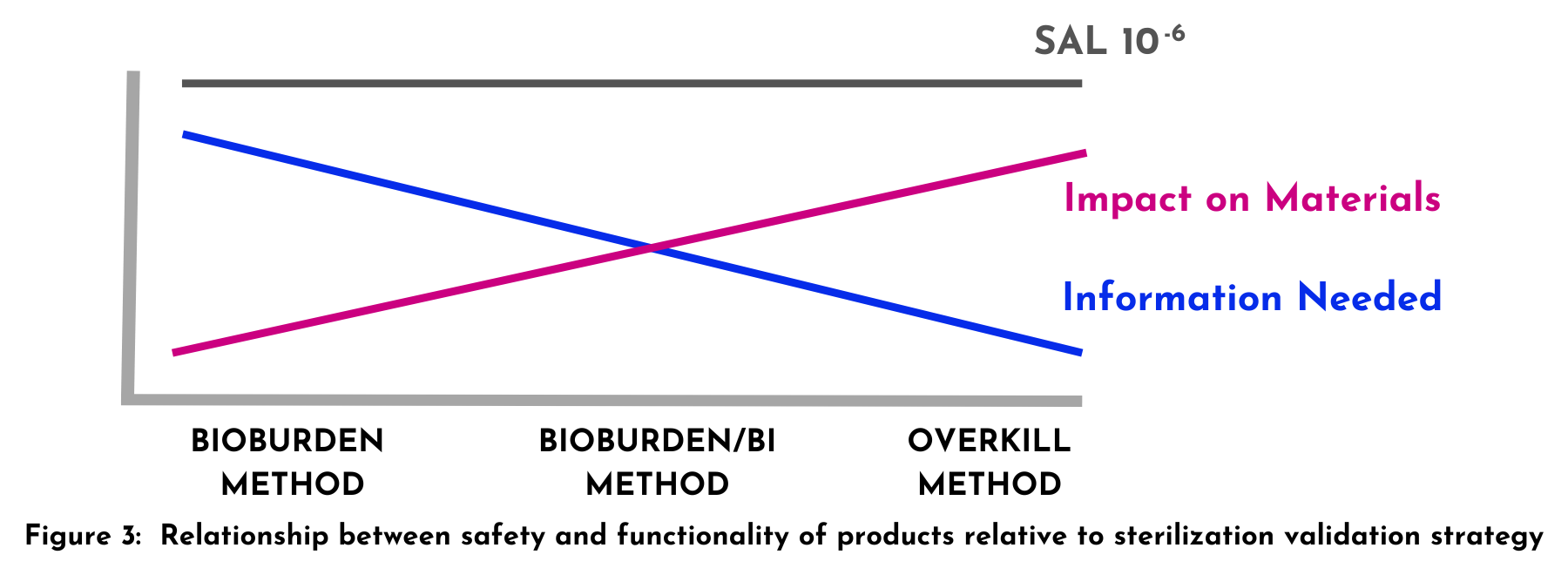
When choosing the appropriate strategy for your product, the safety/functionality relationship must be kept in mind. Figure 3 provides a visual of this relationship and will help in your evaluation.
How to Perform Sterilization Validations
Sterilization validations are confirmation of the defined and desired sterility assurance levels (SALs).2
As mentioned by (name), ISO standards are available for traditional (referred to as “established”) sterilization methods. Niche sterilization methods (referred to as “novel”) may not have a dedicated ISO standard. In these cases, sponsors often select a more general ISO standard for their sterilization validation study. For example, the supercritical carbon dioxide sterilization method follows ISO 14937.
These sterilization validation studies are often conducted in partnership with Contract Research Organizations (CROs). Depending on capabilities, expertise, and access to necessary equipment, medical device and therapeutics companies may choose to conduct these validation studies in-house.
Sterilization Validation Methods for Medical Devices and Therapeutics
Overkill Method
The overkill method confirms the SAL by using what is considered to be the harshest conditions. This method requires that the hardest-to-kill microorganism be established (“challenge organism”). Once this is determined, you must demonstrate the ability to inactivate >6-logs (1 million-plus) of the challenge organism. Common challenge organisms include Bacillus atrophaeus for ethylene oxide, dry heat, and supercritical carbon dioxide sterilization modalities, and Geobacillus stearothermophilus for steam, vaporized hydrogen peroxide, and supercritical carbon dioxide sterilization modalities. It is based on the idea that the challenge organism is a greater challenge for the sterilization modality in terms of contamination levels and biological resistance when compared to the actual bioburden the product, process, or facility will encounter.
The overkill method has two key advantages:
- It is widely used and well-understood by FDA reviewers.
- It does not require expensive biological monitoring of production processes and manufacturing facilities.
But as with any method, it has its limitations. Overkill may not be the best choice for sensitive products. Since you must double the amount of exposure time to achieve a SAL of 10-6, you could subject your product to an overly aggressive sterilization cycle that compromises functionality.
Bioburden Method
The bioburden method requires routine monitoring of the microbial contaminants at the facility, in the manufacturing process, and on the product. This monitoring requires both quantification of the level of contamination and the identification of the microbial contaminants on an ongoing basis. The sponsor must demonstrate that the selected sterilization modality can effectively inactivate the identified microbial bioburden (quantity and type). While it is infrequently used due to the cost associated with monitoring, this method can be useful for highly sensitive products whose functionality hinges on the preservation of the mechanical and chemical utilities of the material. Despite the higher cost when compared to the overkill method, it is more cost-effective than aseptic manufacturing.
Combined BI/Bioburden
As the name implies, this method combines elements of both the overkill and the bioburden methods to validate the desired SAL for the chosen sterilization process.
In this method, regardless of what the native bioburden is determined to be, the sterilization validation process has to demonstrate the ability to eliminate at least 103 microorganisms. Because of the lower bacterial indicator load, a milder sterilization protocol can be used. This is beneficial when processing products comprised of materials that are sensitive and have limited sterilization modality options.
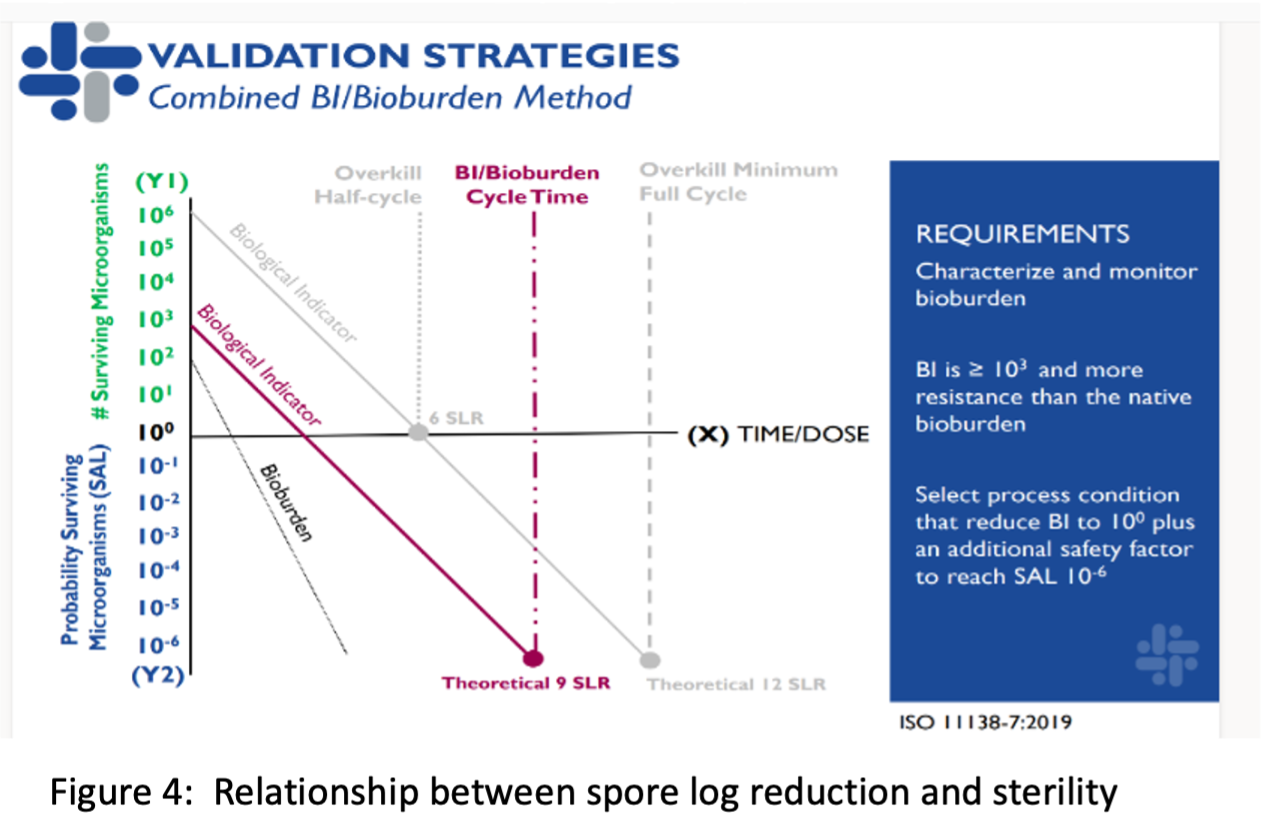
Despite this approach requiring a lower bioburden (at least 103 versus 106), it still requires the same additional 6-log safety factor utilized in the overkill method. The lower bioburden is allowed because environmental microbial monitoring of the facility, process, and product is monitored intermittently. The combined method requires less monitoring than the bioburden method. The combined method is an effective way to use the lowest amount of sterilant to preserve the functionality of sensitive devices while ensuring safety and keeping production costs low by limiting the type and amount of environmental monitoring required. Although this sterilization validation method was rarely used historically, it is being more frequently utilized as the FDA becomes more comfortable with the approach.
Understanding the difference between SAL and log reduction is important because the log reduction, not SAL, sets the three methods apart as shown in Figure 4. While SAL measures the probability of organisms surviving the sterilization process, log reduction measures the percentage of live microorganisms eliminated during sterilization.3
NovaSterilis: Your partner in sterilization validation
Do you need assistance with validating your chosen sterilization method for a new product or a change in sterilization modalities? NovaSterilis can be a resource to help you. We can develop and/or support your sterilization validation needs as part of your global regulatory strategies. We also have the equipment, process expertise, and infrastructure required to conduct optimization processes, load configuration, and sterilization studies tailored to your unique requirements. Our team has extensive experience with both the overkill and the combined BI/bioburden validation methods.
Schedule a Consultation to Learn More.

References:
- WHO. Sterility testing. Available: https://www.who.int/teams/health-product-policy-and-standards/standards-and-specifications/sterility-testing
- STANDARD, BRITISH, and BSEN ISO. “Sterilization of health care products—Moist heat—.” (2006). Available: https://www.iso.org/obp/ui/#iso:std:iso:17665:-1:ed-1:v1:en
- Sandle, Tim. Pharmaceutical microbiology: essentials for quality assurance and quality control. Woodhead Publishing, 2015. Available: https://www.sciencedirect.com/science/article/abs/pii/B9780081000229000128?via%3Dihub